General Outline for the Production of Homologously Recombined ES Cells
- Client consults with Core Manager: ekothari@ucsd.edu
- Client designs targeting plasmid and screening strategy
- Client creates targeting plasmid
- Client confirms practicality of screening strategy and provides it to Core
- Client purifies DNA according to Core protocol
- Core prepares ES cells and electroporates DNA
- Core cultures and drug selects ES cells
- Core picks ES cell clones
- Client prepares & screens ES cell DNA
- Core chromosome counts positive clones & prepares cells for injection
General Outline for the Injection of Blastocysts to Generate Knockout Mice
- Client or core provides prepared ES cells (client provided cells require mycoplasma test results)
- Core prepares blastocysts
- Core injects 1 or 2 clones of ES cells
- Core implants injected blastocysts into foster mice
- Core keeps resulting pups until coat color emerges
- Client screens chimerics
- Client breeds chimerics for germ-line transmission
For a more detailed description of the targeted homologous recombinant process, please refer to our Practical Guide below.
Practical Guide to Gene Targeting and
Creating a Knockout Mouse

Overview of the Creation of a Conventional Knockout Mouse
The client provides the Core with a linearized targeting vector that contains a null or mutated gene of interest. The Core performs the electroporation, drug selection, subclone isolation, cell growth, and freezing of subcloned ES cells. A duplicate sample of each subcloned cell line is kept by the Core as a master plate frozen at -80o C, while a sample of each subcloned cell line is returned to the Client. The Client purifies DNA from each subclone and screens for appropriate gene recombination. The client informs the Core regarding which subcloned cell lines are homologous recombinants. The Core thaws and expands those clones, makes liquid nitrogen stocks of those cells, and returns another culture of each cell line for the Client to reconfirm by screening and further characterization. Lastly, the Core counts the chromosomes for the clones and the best two clones are prepared for Blastocyst injection. Chimeric mice are generated and transferred to the Client's colony. The Client sets up breeding of chimeras to allow germline transmission of the knockout allele by generating complete heterozygous agouti mice. These mice are then bred together to obtain homozygous knockout mice.
This service uses some of the most complex technology of any offered by a core facility, and may be subject to many unforeseeable variables. The variables that affect performance are discussed below. Key points to be addressed are denoted by the circled numerals in the figure at left.
1. Generation of a Targeting Construct (Investigator Responsibility)
Targeting to generate a null or mutated allele is usually accomplished by insertion of a selectable marker (usually neomycin) into a gene causing disruption of splicing, promoter function, or reading frame, with or without deletion of some of the gene. Incorporation of the altered gene into the mouse genome depends upon replacement of the endogenous gene by homologous recombination through both of the arms of the altered gene into one allele of genomic DNA. As starting material, a genomic clone of reasonable length must be obtained from a 129/SvJ mouse library. For adequate frequencies of homologous recombination in our facility, it is recommended that there be at least 4 Kb of uninterrupted sequence on one arm with 2 Kb on the other. This is ideal. We have worked with less homology. It is not that fragments of sub-optimal length will not work, they will simply work at a much lower efficiency than we allow for in costing out the time and materials for an individual transfection. If the efficiency is far less than 1%, we will clearly find little or no homologous recombination in the 100-400 clones that we pick per electroporation.
For investigators new at the creation of these targeting vectors, we can refer you to UCSD labs that have a number of cloning vectors, including pgk-neo, the most commonly used. It is important that the cloned DNA be from the same mouse strain as the ES cells, which in the case of this facility (and most) is 129/SvJ. This 129/SvJ mouse genomic library is available from Stratagene. The use of non-isogenic DNA is not recommended to target the 129 ES cells because there will commonly be a substantial reduction in the efficiency of homologous recombination due to sequence divergence from strain to strain. It may even be that substrain variation in 129 should be considered; this has yet to be determined.
Some investigators like to include the thymidine kinase gene (TK) at the distal end of the genomic DNA of their constructs ("double selection"). This permits selection against the presence of TK in the genome, using gancyclovir simultaneously with selection for G418 (neo) resistance. Clones positive for TK cannot have undergone a correct homologous recombination event. Ideally, survivors of gancyclovir selection are enriched for homologous recombinants. Diptheria toxin (DT) has also been used at times, and its presence self-selects cells without the addition of any selection drugs. However, it has become apparent that the degree of enrichment obtained is highly variable, and with a targeting vector containing the lengths of homology recommended above, is unlikely to be above two fold. Gancyclovir is also associated with some toxicity in some hands. Please inform us if you choose to do so.
Those not wishing to tackle their own targeting vector construction may choose to contract out the task to a commercial operation (e.g., Genome Systems Inc).
With an adequate targeting vector within the optimal size range, a reasonable expectation is that 0-10% of selection-resistant clones picked will have undergone homologous recombination. Published rates of homologous recombination from adequate constructs vary from 0.3% to 50%, for reasons which are poorly understood. Similar ranges are seen in our facility.
Scheduling an Appointment
Please contact ekothari@ucsd.edu to set up all appointments for the initial meeting to discuss construct design and potential screening strategy and subsequent meeting to verify screening strategy and handing over completed paperwork and DNA preparation according to the Core's protocol. Prior to the initial meeting, the Client must complete the Transgenic Core's Request for Services Form available here.
2. Electroporation of ES Cells (Core Responsibility)
In our facility, we use R1 embryonic stem cells derived from 129 SvJ mice for transfection. Cloned DNA isogenic with the 129 SvJ strain is supplied by investigator (200 µg see protocol for core purification procedure) and is electroporated by the Core into stem cells under predetermined conditions. Thirty million cells are plated onto three 15 cm culture dishes in the presence of embryonic feeder cells and Lif. After 10 days of drug selection, between 100-500 clones will be seen on the plates. A subset of these will be chosen by morphologic criteria for picking, expansion, freezing and submission to investigator for DNA analysis.
The number and quality of clones that grow will be dependent, in part, on the quality of the electroporated DNA. To try to reduce this variable, we request that investigators consult us for optimal methods for preparing their DNA. The electroporation conditions and the quality of the ES cell culture conditions will also affect the number of clones. We have empirically optimized both of these conditions and use the same for all targeting vectors. Additionally, two controls are run with every transfection. The first control is cells electroporated with a Neo-resistant genetic marker. Getting a consistent number of Neo-resistant colonies after the mandatory drug selection period indicates that the conditions for electroporation procedure were optimal. The second control is carried out to gauge the level of purity of DNA provided by investigator. Cells are transfected with control DNA, purified by the Core's recommended purification method. The resulting Neo-resistant colonies are expected to have a consistently high ratio of excellent/poor morphology.
There are two principal factors that influence the proportion of homologous recombinant clones in those that grow under selection. These are the length of homology of the targeting vector and the accessibility of the targeted locus. The latter variable is entirely unpredictable, but the length of homology in each arm of the targeting vector is under the control of the investigator and should be within the recommended boundaries. Whether other factors might influence efficiency has yet to be determined.
We select clones for expansion that appear to be undifferentiated and well shaped, and hopefully, most likely to contribute to the germline of any subsequent chimeric animals. The morphologic criteria used are valuable for excluding clones that are unlikely to contribute to the germline, but they are not adequate to determine with certainty which clones will contribute to the germline. Before we start picking clones for the given transfection, we compare the morphologies of the resulting clones to be picked with the our second transfection control. If the Core finds that the investigator's clones have poor morphology, the investigator will be invited to the ES lab to view the controls and compare them to their experiment. We will select the best clones in any transfection, aiming for 100-400, and they will be retained in the facility as frozen master plates. Two 96-well duplicate plates of each master plate will be returned to the investigator for further analysis. Optimally, the master plates must be thawed no more than one month after freezing, so the client should screen the subclones as quickly as possible. Colonies that are frozen longer than one month will most likely be more difficult to recover, and their viability and potential capability will be compromised.
3. Screening Strategy
Southern blotting is the screening method recommended by the Core for the most complete and definitive analysis of your clones. You must produce a detailed restriction map of your genomic clone in order to identify restriction sites that will be crucial for the screening strategy. The most important point is to make a probe that will hybridize to an endogenous sequence that lies completely outside of any sequences contained in the targeting vector. To do this you must identify one restriction site that lies outside of any sequence contained in the targeting vector (the other can be either inside or outside). Then, have your probe hybridize to a short sequence contained within this restriction fragment but completely outside of any sequence in your targeting vector. This is the only way to distinguish a homologous recombinant from a random integrant. Usually an additional restriction site is found in the neo gene that has been inserted so that there is a large enough "shorter" fragment generated by digestion in a homologous recombinant. (There should be a size difference in the fragments of around 2 kb.) This is then seen in the bands on a Southern blot, between wild-type sequence and a homologous recombinant. Alternatively, the insertion of the neo gene (~2.5 kb) will cause a supershift in the homologous recombinant versus the wild type. The probe should be a fragment of between 300 bp and 1 kb that produces a sensitive and specific band on a Southern Blot of 5 µg genomic DNA per clone.
Because the ES cells can survive for only a short time stored in master plates at -80o C, the Core requires evidence from the client prior to transfection that they can quickly and effectively screen the clones they will receive. Therefore, the client should perform a pilot experiment with wild type R1 ES genomic DNA (which can be provided by the Core) to demonstrate the specificity and sensitivity of the probe before the transfection can be scheduled.
The duplicate 96-well plates provided by the ES facility provide enough DNA for two Southern blots and a number of PCR reactions. The strategy for identifying the recombinants should be reliable enough for this to be adequate material. We cannot stress enough that you must have a probe specific enough to detect any homologous recombination event and a construct designed so that there is a diagnostic Southern band. Keep in mind that the recombination occurs only on one of the two chromosomes and even your positive clones will still have the wild-type banding pattern. The ES cells are male, thus genes found on the X or Y chromosome will be represented only once.
With appropriately prepared DNA, it is a reasonable expectation that the ES facility will pick up 100-400 clones for analysis, if that many clones grow. If a transfection yields lower numbers of clones, the clones obtained will be analyzed before performing a repeat transfection. If some of the clones obtained are homologous recombinants, it is unnecessary to repeat an already successful transfection. The hope is that each transfection will produce at least 3 positive clones after final analysis.
4. Blastocyst Injection (Core Responsibility)
Having identified clones in which an homologous recombination event has occurred we will thaw and expand the counterparts in our facility and freeze these few chosen clones in liquid nitrogen. Then we will provide larger cell samples for the investigator to confirm that these are indeed the clones that they are interested in. We will then prepare the cells for injection into mouse blastocysts. We inject into a C57Bl/6 mouse strain, and purchase superovulated, mated mice from a commercial supplier. We inject into blastocysts retrieved from a super ovulated mated C57Bl/6 mouse strain. Per targeting vector, we aim to inject 30 to 40 blastocysts for each of two clones with 8-10 ES cells per blastocyst.
5. Injected Embryos
Injected embryos are implanted into pseudo-pregnant females, and pups are born after a 17-day gestation period. Chimeric mice can be identified by coat color 10-14 days after birth. In cases where few pups are born or very little chimerism is seen, we will select further clones and inject them as needed.
6. Mating (Investigator Responsibility)
At approximately 2 weeks of age, chimeras will be shipped from the Core to the Investigator to be weaned at 3 weeks old, then bred at 6 weeks. Matings should be set up in order to determine germline transmission. As a general guide as to which mice to mate, most germline chimeras will be males because the original 129 ES line is a male cell line, but one is advised to mate all good chimeras because occasionally, germline chimeras have been reported in female lines too. With the ES cell (129) and host (B6) combination used in this facility, it is prudent to mate all chimeras that you receive to ensure you get germline transmission. We have found, however, that those less than 30% chimeric are less likely to go germline. Black mice are used for mating, either C57 Bl/6 or Black Swiss, and germline transmission is detected by agouti (versus black) coat color in the pups. Black Swiss, a non-inbred strain, are generally preferable for breeding as they are cheaper and more fecund than the Bl/6. Both strains are available from Harlan Sprague Dawley, Jackson and Charles River. Matings are best set up with age matched females, 2-3 females per chimera. Once the germline chimeras have been identified, Investigators should breed into the genetic background that is of interest to them. The most straightforward method is to keep everything on a 129 background, bearing in mind that there are many sub-strains of 129.
There may be breeding problems. Even a high percentage chimera may fail to go germline, which is nothing more or less than bad luck. You may even have germline transmission, but only of the non-targeted allele. Sometimes the chimeras are hermaphrodites, or they may be infertile for no detectable reason. A fertile chimera should have fathered 80-100 non-germline pups before being discarded as non-productive, although it is usual that a germline transmitter will become evident in the first few litters. Once a single germ-line offspring containing the altered allele is identified, this animal should be bred since it is a pure heterozygote and not a genetic mosaic or chimera. No further breeding of the original chimeras is necessary.
It is the aim of the facility that if all guidelines have been followed, the investigator will succeed with chimeras produced from two different clones giving germline transmission. If this goal is not accomplished, we will work with the investigator to do the best that we can to achieve it.
A Timeline for Knockouts after DNA is Provided to the Core
Step in the Process
|
Responsibility
|
Timeframe
|
| 1. Wait for Transfection Availability |
|
1-2 weeks |
| 2. Transfection, Picking, Expanding, Freezing |
CORE |
4 weeks |
| 3. Screening |
Investigator |
1 month max |
| 4. Expanding Homologous Recombinant Clones |
CORE |
3 weeks |
| 5. Reconfirmation |
Investigator |
2 weeks |
| 6. Karyotype |
CORE |
2-3 weeks |
| 7. Blastocyst Injection |
CORE |
1-2 weeks |
| 8. Transfer Chimeric Mice after Blastocyst Injection |
CORE |
5 weeks |
| 9. Breeding for Germline Transmission |
Investigator |
1-6 months |
| 10. Breeding to Homozygosity |
Investigator |
2-3 months |
| Reasonable Expectation |
|
1 year |
Conditional Knockout Mice Using the Cre-loxP System
When global removal of a gene of interest using conventional knockout methods results in embryonic lethality, investigators can choose to produce a conditional knockout line of mice where tissue-specific deletions can still be studied in vivo. To knock out a gene in a specific tissue, the investigator must subclone an important exon in between two loxP sites (i.e. clone loxP elements in each of the introns flanking the exon to be deleted). Homologous recombinants are generated by a homologous recombination replacement event as with conventional targetings. The difference is that the gene will retain normal function in the ES cells and in the parental chimeras and agouti heterozygotes and homozygotes, thus allowing the mice to survive and breed. Then, after germline transmission has occurred and the mice are bred to homozygosity, the "floxed" mice are bred to a tissue-specific Cre transgenic mouse, and in the resulting offspring, the exon will be removed only in that tissue.
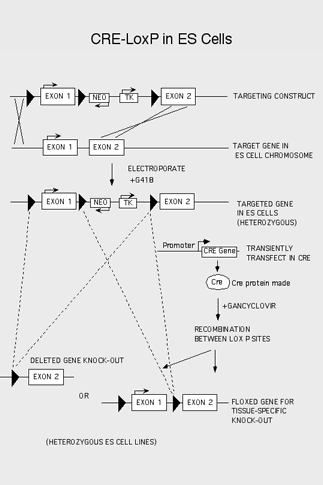
Making a conditional knockout mouse requires nearly twice as much time and work to complete successfully. The investigator must use the vector that contains three loxP sites, two of which flank the important exon, and the 3rd of which lies just 3’ of the Neo and TK selection markers (see diagram). The first transfection follows the same procedure as for conventional targetings. After the original homologous recombinants are generated (called parental clones), they are expanded, reconfirmed, and chromosome counted as with the conventional targeting. Then the Core performs a second, transient, transfection, with a plasmid containing the gene for the Cre enzyme. The cells are placed under gancyclovir selection (which kills TK expressing cells) to remove parental cells and select for those cells that have undergone recombination to generate type I and type II deletions. These cells are then screened, expanded, reconfirmed, chromosome counted and finally injected to make the "floxed" mice.
The investigator must design a screening strategy that will enable a definitive analysis of all clones by distinguishing between wild type, parentals, type I, and type II deletions (see diagram). Again either PCR or Southern analysis is sufficient, but both are recommended for complete verification.
Recommended Reading
- Robertson EJ: Teratocarcinomas and Embryonic Stem Cells: A Practical Approach (IRL Press 1987)
- Joyner AL: Gene Targeting: A Practical Approach (IRL Press 1993)
- Hogan B, Costantini F and Lacy E: Manipulating the Mouse Embryo (CSH, 1994)